
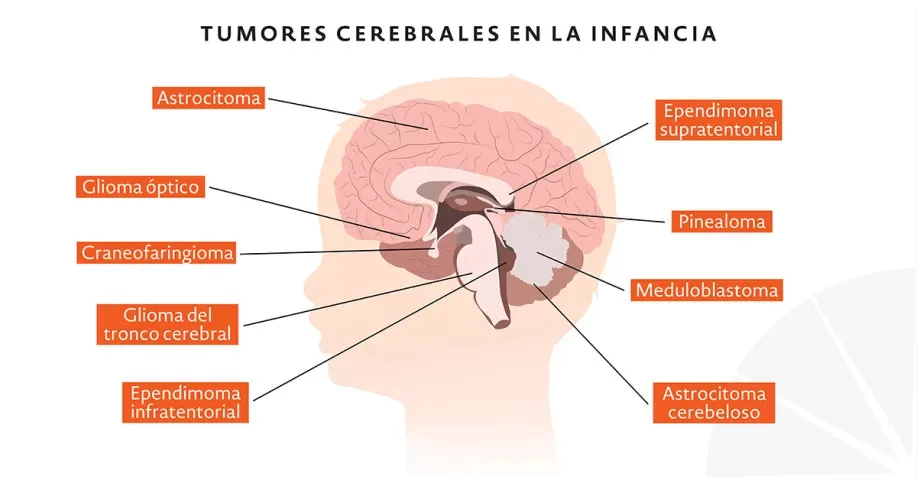
Los tumores del sistema nervioso central en niños presentan una incidencia de 2.76 a 4.03/100,000 niños al año, hecho que nos hace entender que a pesar de ser enfermedades poco frecuentes si son dignas de ser tomadas en cuenta. Se diferencian de los que aparecen en edad adulta no solo en la histología, es decir su composición celular, sino también en la presentación clínica, porque ésta última en los niños, depende de la localización del tumor y de la edad en la que se presenta.
Este tipo de tumores se produce a expensas del tejido de sostén del tejido nervioso y constituyen un grupo significativo de enfermedades identificadas como Gliomas son un grupo heterogéneo de tumores, que en la mayoría de los casos no sufren transformación maligna y ocasionalmente pueden tener una regresión espontanea. Pueden recurrir aun si fueron resecados completamente. En un porcentaje variable desde su inicio no son tumores benignos y pueden afectar de manera negativa la calidad de vida, aun en los pacientes con astrocitomas pilocíticos en cerebelo que son tratados solo con cirugía. Los pacientes con este tipo de tumores, al tener afectaciones del tejido nervioso como efecto paralelo durante su crecimiento están en riesgo de presentar alteraciones adaptativas, cognitivas, así como del lenguaje, memoria, atención y problemas de función especial. Se ha estimado que en conjunto constituyen el 30 a 50% de los tumores del sistema nervioso central en niños. Los gliomas más comunes en niños son el astrocitoma pilocítico y el astrocitoma fibrilar (los tumores astrociticos se definen colectivamente como tumores derivados de los astrocitos). Los Gliomas de alto grado a su vez representan cerca del 10% de todos los tumores cerebrales pediátricos. Se comportan agresivamente dando manifestaciones relacionadas con su efecto principal de crecimiento y como consecuencia de la invasión local, esto hace que su pronóstico sea pobre a pesar de todos los tratamientos disponibles. Se presentan con una distribución semejante en hombres y mujeres. Cerca de un tercio se originan en la región supratentorial (30-50% en los hemisferios cerebrales). Se presentan con mayor frecuencia en el grupo de edad de 15 a 19 años. Los tipos histológicos más comunes son el astrocitoma anaplásico (OMS grado 3) y el glioblastoma multiforme (OMS grado 4). En el caso de los tumores malignos de estirpe astrocitica se sabe que es un tumor embrionario altamente maligno con tendencia a la diseminación leptomeningea, en algunas series se reporta como el tumor cerebral maligno más común en niños. El 20-30% de los estos tumores se presentan en la primera década de la vida con un pico a los 5 años y un leve predominio por el género masculino; histológicamente es un tumor neuroectodérmico primitivo indiferenciado que se origina en cerebelo y puede conservar caracteres primitivos de los pilocitos. Se ha propuesto que la exposición previa a tratamiento con radioterapia craneal (leucemias agudas, linfomas y otros tumores), aumenta el riesgo de desarrollar un glioma de alto grado (1 a 3%), con una relación directa de dosis-efecto y con un período de latencia documentado de 9 a 12 años posterior a la exposición, por lo que se recomienda realizar vigilancia clínica estrecha de forma regular, que incluya valoración oftalmológica y neurológica; durante un período mínimo de 10 años posterior a la exposición a tratamiento con radionúclidos. Los niños que presentan algunos síndromes raros, con anormalidades que afectan la regulación de la proliferación celular y apoptosis, tienen un mayor riesgo de presentar gliomas de alto grado y en particular el meduloblastoma estas enfermedades se han visto asociadas con síndromes como:
• Li-Fraumeni (defecto en el gen TP53) • Neurofibromatosis tipo I
• Turcott (también mayor riesgo para cáncer colo-rectal, meduloblastoma y ependimoma) • Síndrome de Gorlin
• Esclerosis tuberosa
Los pacientes con presencia de cualquiera de los síndromes mencionados deberán recibir una vigilancia clínica estrecha, incluyendo valoración oftalmológica y neurológica de forma regular se recomienda que el intervalo de tiempo, para la valoración clínica neurológica, en los pacientes con factores de riesgo para el desarrollo de tumores cerebrales, sea de 6 a 12 meses. Ante la sospecha clínica de presencia de tumor en sistema nervioso central, enviar al paciente a una valoración especializada a la brevedad posible, para manejo integral oportuno y limitación de daño. El cuadro clínico depende de la localización, histología y edad del paciente. Los tumores de bajo grado de malignidad tienen una evolución insidiosa mayor a 6 meses y los tumores de alto grado suelen dar manifestaciones en un período más corto. La Resonancia Magnética (RM) simple y con gadolinio, es el estudio de imagen recomendado para la evaluación de los niños con sospecha de tumor cerebral, ya que nos da un diagnóstico preoperatorio probable, permite planear el tratamiento y sirve de guía para el seguimiento de los pacientes. Ante cualquier paciente con alteración neurológica con o sin síntomas, se debe realizar un estudio de imagen con RM de cráneo (simple y con gadolinio) En caso de no contar con este recurso, tomar tomografía de cráneo simple y contrastada; como abordaje inicial En pacientes con tumores cerebrales de difícil acceso quirúrgico, ó en quienes la resección completa no esta recomendada, se debe realizar la biopsia por estereotaxia, para valorar el uso de radioterapia y/ó el tratamiento quirúrgico. Algunos autores recomiendan que, en niños con astrocitoma de bajo grado, la segunda resección se posponga hasta que haya una progresión detectada por síntomas clínicos o neuroimagen. En casos con progresión del tumor y donde no es factible una segunda resección, podría considerarse la radioterapia en niños mayores de 3 años, en un intento de fines curativos. El manejo inicial de los gliomas de alto grado gira alrededor de la estabilización del paciente, la descompresión intracraneana y la posibilidad de toma de biopsia y/o resección quirúrgica completa La evaluación inicial de un tumor cerebral y sobre todo si es glioma de alto grado, debe ser realizada por un neurocirujano, idealmente con experiencia en neurocirugía pediátrica. La conducta quirúrgica La conducta quirúrgica La conducta quirúrgica La conducta quirúrgica en un glioma de alto grado siempre que sea posible, será resección radical (igual o mayor a 90%) Se debe realizar estudio histopatológico que aborde características como pleomorfismo nuclear*, mitosis*, necrosis*, proliferación endotelial*, y de ser posible establecer la presencia de marcadores como Ki-67 o MIB-1. En el tratamiento de gliomas de alto grado supratentoriales, en niños mayores de 3 años, se recomienda utilizar radioterapia conformal con margen pequeño, a dosis de 50 a 60 Gy combinada con quimioterapia, posterior a cirugía. Existe evidencia de mejoría de la sobrevida global en niños con gliomas de alto grado, sobre todo glioblastoma, con esquemas de quimioterapia a base de lomustina, prednisona y vincristina por lo que se recomienda el uso de nitrosureas (lomustina) en el esquema de quimioterapia convencional para pacientes con gliomas de alto grado En el campo de la medicina y especialmente en el área de oncología, al hacer referencia a los astrocitoma, se identifican como toda aquella neoplasia o tumor formada principalmente por astrocitos, uno de los principales tipos de células gliales que alimentan y dan soporte a las neuronas. Dicho tumor es una masa de astrocitos que se produce ante un crecimiento y proliferación anómala, patológica y descontrolada de uno de los tipos de tejido glial presentes en el sistema nervioso, tratándose de uno de los principales tipos de tumor cerebral. Los tumores grado I tienen bajo potencial proliferativo y tienen
posibilidad de cura al ser resecados quirúrgicamente. Los tumores grado II son tumores infiltrantes, pero de baja actividad proliferativa celular, tienden a recurrir y en algunos casos, como los gliomas, a progresar a grados superiores (III y IV). Los tumores grado III son lesiones con evidencia histológica de malignidad y los grado IV tienen evidencia de malignidad citológica con predisposición a necrosis y están relacionados con una evolución rápida y fatal de la enfermedad, como lo es el glioblastoma. Estos tumores grado III y IV son denominados de “alto grado” o “malignos”.
El astrocitoma fibrilar es un tipo de tumor cerebral que se origina en los astrocitos, un tipo de célula glial del cerebro. Estos tumores se clasifican según su apariencia y comportamiento. Existen diferentes formas de astrocitoma fibrilar, cada una con sus propias características y posibles implicaciones para el tratamiento y el pronóstico. Estas formas pueden variar en términos de agresividad, tasa de crecimiento y respuesta al tratamiento. Comprender el tipo específico de astrocitoma fibrilar es crucial para determinar el enfoque de tratamiento más adecuado. Astrocitoma fibrilar difuso: Un tipo común de astrocitoma compuesto de astrocitos fibrilares, que a menudo se encuentran en el cerebro y la médula espinal. Astrocitoma fibrilar anaplásico: Una forma más agresiva de astrocitoma fibrilar caracterizada por un crecimiento rápido y mayores probabilidades de recurrencia. Astrocitoma fibrilar pilocítico: Un tipo de astrocitoma fibrilar de crecimiento lento que generalmente se presenta en niños y adultos jóvenes, con un mejor pronóstico en comparación con otros tipos. Astrocitoma fibrilar pleomórfico: Una variante rara y de alto grado de astrocitoma fibrilar, caracterizada por astrocitos pleomórficos anormales, que a menudo requiere un tratamiento agresivo. Astrocitoma fibrilar subependimario: Un tipo de astrocitoma fibrilar que surge en la pared de los ventrículos laterales del cerebro y que generalmente se encuentra en personas con esclerosis tuberosa.
El astrocitoma fibrilar es un tipo de tumor cerebral que puede presentarse tanto en niños como en adultos. Existen varios factores que pueden aumentar el riesgo de desarrollar esta afección. Comprender estos factores de riesgo puede ayudar a las personas y a los proveedores de atención médica a identificar posibles problemas y tomar las medidas adecuadas. El astrocitoma fibrilar se suele diagnosticar mediante diversas pruebas y exploraciones por técnicas de imágen que ayudan a evaluar el cerebro para identificar cualquier anomalía. Estos procedimientos de diagnóstico proporcionan información valiosa a los profesionales de la salud, lo que les permite realizar un diagnóstico preciso. El proceso implica una combinación de evaluación de la historia clínica, examen físico y pruebas especializadas para confirmar la presencia de un astrocitoma fibrilar. Si sospecha que tiene algún síntoma relacionado con esta afección, consulte con un proveedor de atención médica de inmediato para que le realice una evaluación y un diagnóstico exhaustivos.
Pruebas de imagen: Resonancia magnética y tomografía computarizada Biopsia: Examen de muestra de tejido
Exámenes neurológicos: Evaluación de la función cerebral
Prueba genética: Identificación de mutaciones específicas
Análisis de sangre: Comprobación de biomarcadores Punción lumbar: Análisis del líquido cefalorraquídeo Tratamiento del astrocitoma fibrilar
El tratamiento del astrocitoma fibrilar suele implicar una combinación de cirugía, radioterapia y quimioterapia. El enfoque específico depende de factores como el tamaño del tumor, la ubicación y el grado. La cirugía suele ser el primer paso para extirpar la mayor parte posible del tumor. La radioterapia se puede utilizar después de la cirugía para atacar las células cancerosas restantes. También se puede considerar la quimioterapia, especialmente para tumores que son difíciles de tratar quirúrgicamente o con radiación. Además, el control y la atención de seguimiento continuos son esenciales para realizar un seguimiento de la respuesta del tumor al tratamiento y controlar los posibles efectos secundarios.
Cirugía: El tratamiento principal del astrocitoma fibrilar implica la extirpación quirúrgica del tumor para reducir la presión sobre el cerebro y evitar un mayor crecimiento.
Terapia de radiación: Se puede utilizar radioterapia después de la cirugía para atacar las células cancerosas restantes y reducir el riesgo de recurrencia.
Quimioterapia: En algunos casos, se puede recomendar quimioterapia para retardar el crecimiento del tumor o tratar el astrocitoma fibrilar recurrente.
Terapia Dirigida: Los medicamentos de terapia dirigida se pueden utilizar para atacar específicamente las células cancerosas y, al mismo tiempo, minimizar el daño a las células sanas.
Ensayos clínicos: La participación en ensayos clínicos puede ofrecer acceso a tratamientos y terapias innovadores que pueden ayudar a mejorar los resultados de los pacientes con astrocitoma fibrilar.









