Dr. Guillermo Flores Flores. [email protected], [email protected]
Sarcoma Sinovial
Los sarcomas de tejidos blandos son tumores malignos originados de una porción especializada del mesodermo, una de las capas embrionarias que se forman después de la implantación del producto de la concepción en el ser humano, y en particular de un tejido especializado conocido como mesénquima, este mesénquima produce por lo tanto los derivados mesenquimatosos que suelen ser en cuanto a diferenciación distintos de los derivados del ectodermo y del endodermo que suelen ser de origen epitelial y cuya representante más conocido es la piel.
Por lo tanto, podemos decir que los tumores malignos de origen mesenquimatoso, en el contexto que nos interesa darán origen a tumores malignos no epiteliales del tejido extraesquelético del cuerpo, que incluyen el músculo, la grasa, el tejido fibroso, los vasos y el sistema nervioso periférico. Ellos forman un grupo heterogéneo de neoplasias mesenquimatosas, ahora si entendemos porque se denominan mesenquimatosas, que se clasifican en una base histológica de acuerdo con el tejido adulto al que se parece o se supone que derivan.
Estos tumores son poco frecuentes si los comparamos con los tumores más frecuentes del cuerpo humano como es el caso del cáncer de mama en el cual se presentan en promedio 14 casos por 100,000 habitantes en todas las poblaciones. Los tumores mesenquimatosos tienen una incidencia anual de alrededor de 2-3/100 000, suponen menos del 1% de todos los tumores malignos y el 2 % de todas las muertes relacionadas con cáncer, aunque en los últimos años, los sarcomas de tejidos blandos representan el 8% de todas las neoplasias malignas.
El sarcoma sinovial (SS) es un tumor maligno de partes blandas perteneciente a la familia de los tumores mesenquimales, de origen incierto, que afecta con mayor frecuencia a adolescentes y adultos jóvenes, y muestra una especial selectividad de vecindad con las articulaciones. El pico de incidencia del sarcoma sinovial se observa en la tercera década (aproximadamente el 30% de los casos ocurren en pacientes de menos de veinte años de edad), y los hombres son más afectados que las mujeres (razón hombre / mujer alrededor de 1,2:1). A pesar de su nombre, el sarcoma sinovial no surge a partir de tejido sinovial. Como con la mayoría de los sarcomas de tejidos blandos, la patogénesis del Sarcoma Sinovial todavía se desconoce y no están bien establecidos los factores de riesgo. Los Sarcomas Sinoviales se clasifican en función de su aspecto morfológico en:
-Bifásicos – Monofásicos – Monofásicos epiteliales (excepcional) – Pobremente diferenciados
El sarcoma sinovial bifásico muestra tanto células fusiformes como células con diferenciación epitelial, en proporción variable. El sarcoma monofásico muestra solo el componente de células fusiformes. El sarcoma monofásico puramente epitelial glandular es una entidad teórica y requiere de la genética molecular para distinguirlo del adenocarcinoma. Los sarcomas pobremente diferenciados muestran uno de los tres patrones morfológicos: células grandes / epitelioides / patrón rabdoide, un patrón de células pequeñas y un patrón de alto grado de células fusiformes, con un comportamiento más agresivo y un mayor porcentaje de metástasis.
Dada la aparente existencia de una similitud microscópica con la sinovial normal, esta entidad fue denominada en 1927, por Smith, con el término de sinovioma, y en 1936, Knox, observando el comportamiento agresivo de una neoplasia francamente maligna, sugirió el nombre de sarcoma sinovial, aunque nunca se ha demostrado totalmente su origen a partir de tejidos sinoviales preformados.
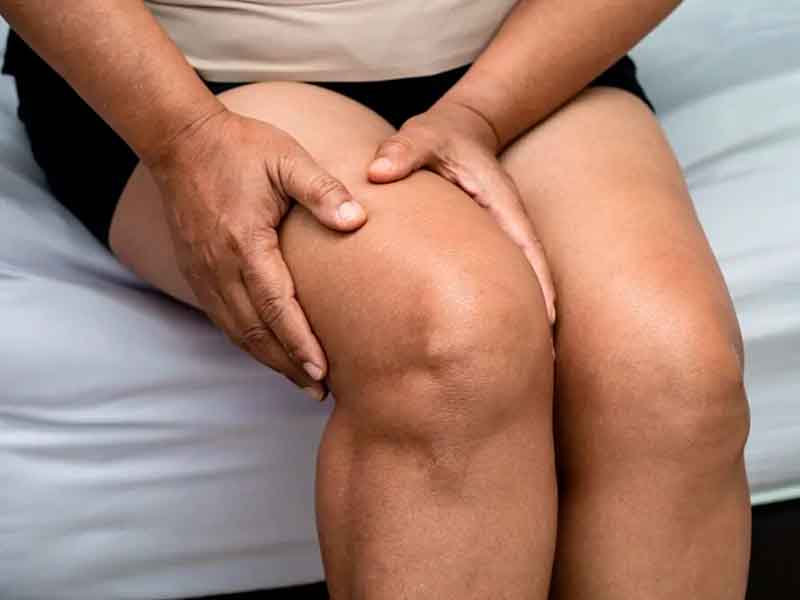
Algunos estudios recientes proponen considerar más apropiadamente a esta neoplasia como un carcinosarcoma, en base a los componentes histológicos descritos es decir componentes epiteliales y componentes mesenquimales. Este es un fenómeno que ocurre en otras partes del cuerpo como es por ejemplo el caso del llamado Tumor Maligno Mulleriano del cuerpo uterino y que en realidad histológicamente constituye otro ejemplo de carcinosarcoma, Desde el punto de vista clínico, el sarcoma sinovial se suele debutar como una masa “palpable profunda, dolorosa” en algo más de la mitad de los pacientes, a veces con limitación de los movimientos, pero rara vez se acompaña de alteraciones funcionales graves y sobre todo que se localiza muy cerca de alguna articulación, un ejemplo frecuente es la articulación de la rodilla.
Otras manifestaciones clínicas particulares están relacionadas con la ubicación del tumor, como la dificultad para deglutir y respirar en los tumores de cabeza y cuello. Generalmente, el tumor crece lenta e insidiosamente, lo que demora con frecuencia el diagnóstico y tratamiento; el paciente refiere molestias desde varios años antes del diagnóstico, y no es raro que se diagnostique erróneamente como sinovitis, bursitis o artritis.
El sarcoma sinovial es más frecuente en adolescentes y adultos jóvenes entre 15 y 35 años. Predominan en las extremidades, y habitualmente se presentan en la vecindad de las grandes articulaciones, especialmente la rodilla. Desde el punto de vista histológico, una forma de presentación común, se distingue un tipo bifásico constituido por un componente de células epiteliales y otro de células fusiformes, y un tipo monofásico en el que predominan las células fusiformes o muy raramente el componente epitelial, estando el patrón bifásico ausente, o solo representa una pequeña porción del tumor.
Asimismo, se reconoce una variante pobremente diferenciada que ofrece una mayor dificultad diagnóstica y puede representar el 20 % de todos los sarcomas sinoviales. Meis-Kindblom propone dividir el grupo de pobremente diferenciados en tres subgrupos: una variante de células grandes, una de células pequeñas y otra de células fusiformes de alto grado. Desde el punto de vista ultraestructural, en el sarcoma sinovial se reconocen elementos con características epiteliales, células fusiformes que semejan fibroblastos y formas intermedias con cúmulos de tonofilamentos, a menudo en una posición paranuclear, microvellosidades irregulares en la superficie celular hacia la luz de los espacios seudoglandulares, uniones celulares de tipo mácula adherente o hemidesmosoma y material de lámina basal en la interface de las células epiteliales y fusiformes.
En la inmunohistoquímica se manifiesta una fuerte positividad a las citoqueratinas en las áreas epiteliales, y a menudo, también en las células fusiformes. Casi todos los sarcomas sinoviales bifásicos muestran, en el componente epitelial, reacción para las citoqueratinas y EMA. En los sarcomas monofásicos, aunque elevado, el porcentaje de positividad para citoqueratinas está algo disminuido, alrededor de 60-70 %, y para los pobremente diferenciados, alrededor del 40 %.
Es importante señalar que las células sinoviales normales o reactivas no expresan citoqueratinas, y aunque otros sarcomas de partes blandas muestran positividad para las citoqueratinas 8 y 18 solo raramente, al contrario del sarcoma sinovial, muestra positividad para queratinas 19 y 7, hecho que puede ser de utilidad para realizar el diagnóstico diferencial con otros tumores. En un porcentaje variable (30-40 %), expresa positividad para la S100 y negatividad para CD34. El sarcoma sinovial también puede mostrar positividad para CD99 (MIC2) y bcl2; este hecho tiene escasa utilidad desde el punto de vista del diagnóstico diferencial, ya que muchas lesiones y tumores de partes blandas expresan el CD99 y bcl2.
El sarcoma sinovial se encuentra asociado, en un 90-95 % de los casos, a una alteración citogenética característica: la translocación cromosómica t(X,18) (P11.2; q 11.2) El punto de ruptura afecta a dos genes SSX (localizados en el cromosoma X) y al SYT (localizado en el cromosoma 18); de la fusión entre el gen SYT y los genes SSX1 o SSX2 resulta la formación de nuevos genes quiméricos: SYT-SSX1 o SYT-SSX2. Los puntos de rotura se han correlacionado con el fenotipo tumoral. Los tumores monofásicos tienen su punto de rotura dentro de la región SSX2 y los tumores bifásicos con un componente epitelial extenso o focal tienen su punto de rotura dentro de la región SSX.
Este tumor, se ha relacionado con un mejor pronóstico: el hallazgo de extensa metaplasia ósea, calcificación o ambas, la presentación en pacientes jóvenes, una prolongada duración de los síntomas antes del diagnóstico (presumiblemente debido a que el tumor sea biológicamente menos agresivo), la localización distal de la tumoración, un tamaño inferior a 5 cm y una actividad mitótica inferior a 10 mitosis por 10 campos de gran aumento.
Este tipo de tumor, aun cuando pueda estar perfectamente caracterizado puede ofrecer dificultades diagnosticas importantes por su localización paraarticular inicial, pero que a medida que pasa el tiempo si se deja sin tratamiento, puede afectar las partes óseas y articulares de las articulaciones anexas a su crecimiento primario y dar la impresión que se trata de un tumor maligno producto de osteoide, es decir en el diagnóstico diferencial de los tumores “grandes” pudiera ser confundido con osteosarcoma.
Por el contrario, se han asociado a peor pronóstico: los tumores mayores de 5 cm, la actividad mitótica superior a 10 mitosis por 10 campos de gran aumento, la existencia tanto de necrosis como de áreas pobremente diferenciadas, especialmente si estas suponen más del 20 % del tumor, la presencia de células rabdoides, un escaso número de mastocitos, un alto grado de atipia nuclear y aneuploidia. Las tasas de sobrevida en cinco años varían de un 25,2 a un 62,5 %; la sobrevida en 10 años varía del 11,2 al 30%.
Estas diferencias entre las tasas de sobrevida en 5 y 10 años reflejan la incidencia relativamente alta de metástasis tardías en pacientes con sarcoma sinovial. El sarcoma sinovial puede recurrir localmente o metastatizar a distancia, especialmente a pulmón y ganglios linfáticos. La incidencia de metástasis ganglionares se ha descrito en un 10 a 15 %. El tratamiento de elección es la escisión local con un amplio margen de tejido sano, para evitar recidivas, asociado a radioterapia, quimioterapia o ambas.
El desarrollo alcanzado por la inmunohistoquímica ha permitido un mayor conocimiento de la histogénesis de los tumores de partes blandas y, a su vez, un mejor entendimiento de la historia natural de la enfermedad que, como puede observarse en los dos casos presentados, existe una marcada diferencia en el comportamiento biológico, el pronóstico y la sobrevida en cada uno de los pacientes.
El incremento en el número de casos permitirá en el futuro poder estandarizar un protocolo de tratamiento, ya no solo para los sarcomas sinoviales como familia, sino para cada uno de los subtipos histológicos de esta enfermedad, lo que tendrá un impacto considerable en la sobrevida de los pacientes.