
Aunque su causa exacta es desconocida, se cree que puede estar relacionada con una respuesta inmunitaria exagerada ante un agente infeccioso en individuos genéticamente predispuestos. La incidencia varía según la región y la etnia, siendo más común en niños asiáticos.
El diagnóstico se basa en criterios clínicos, incluyendo:
- Fiebre persistente por cinco días o más.
- Conjuntivitis bilateral sin exudado.
- Eritema y descamación en labios y cavidad oral.
- Exantema polimorfo en el cuerpo.
- Edema y enrojecimiento en manos y pies.
- Linfadenopatía cervical mayor a 1.5 cm.
Tratamiento - Inmunoglobulina intravenosa (IGIV) para reducir la inflamación y prevenir aneurismas. –
Aspirina en dosis altas durante la fase aguda y dosis bajas en la fase de recuperación. - En casos resistentes, se pueden administrar corticoides o fármacos biológicos.
después de todo lo expuesto podemos decir que la enfermedad de Kawasaki es una afección grave que requiere diagnóstico temprano y tratamiento oportuno para evitar complicaciones cardiovasculares. La investigación continúa para comprender mejor su origen y mejorar las estrategias terapéuticas.
La proporción entre varones y mujeres es de 1,5:1. Se ha observado una mayor incidencia en los meses de invierno y primavera. En población japonesa se ha evidenciado una tasa de recurrencia familiar del 2,1%, con un riesgo relativo de aproximadamente 10 veces comparado con la población general; aproximadamente la mitad de los segundos casos en la misma familia se producen dentro de los 10 días de la enfermedad del primer caso. El riesgo de concordancia en gemelos idénticos es aún más elevado (hasta del 13%). También existe riesgo aumentado a sufrir Enfermedad de Kawasaki entre los hijos de padres que sufrieron Enfermedad de Kawasaki en la infancia. Aunque los hallazgos clínicos, de laboratorio y las características epidemiológicas de la enfermedad sugieren la presencia de un origen o desencadenante infeccioso, hoy en día no se ha podido identificar un agente causal infeccioso único. Tampoco se ha podido demostrar que la enfermedad se asocie a la exposición a fármacos, o que se desarrolle en respuesta a un superantígeno. Una de las teorías actualmente más aceptadas sugiere que la enfermedad puede ser causada por un agente infeccioso que se inhalaría e infectaría células epiteliales bronquiales ciliadas de tamaño mediano. Estudios recientes, basados en el análisis de las grandes epidemias de EK en Japón, sugieren que el agente causal podría ser un agente medioambiental transportado por vientos troposféricos. Paralelamente, la alta incidencia en las comunidades asiáticas y el mayor riesgo entre hermanos de los casos, sugieren que los factores genéticos del huésped son importantes en la patogénesis de la Enfermedad de Kawasaki. Se han publicado algunos estudios de asociación de genoma completo (GWAS) en Enfermedad de Kawasaki y se han identificado varios loci biológicamente implicados en la inflamación, la respuesta inmunitaria y el estado cardiovascular. Así, una hipótesis razonablemente abierta, es que la EK está causada por un agente infeccioso aún por identificar, que produce enfermedad solo en individuos genéticamente predispuestos, particularmente asiáticos. Su rareza en los primeros meses de vida y en adultos sugiere un agente al que estos últimos son inmunes y del cual los lactantes muy pequeños están protegidos por anticuerpos maternos pasivos.
El curso evolutivo se divide en tres fases:
• Periodo febril agudo, que dura aproximada mente 10 días.
• Periodo subagudo, que dura entre 2 y 4 semanas.
- Fase de convalecencia, en que se resuelven la mayoría de los síntomas.
La Enfermedad de Kawasaki presenta las siguientes manifestaciones:
• Fiebre: aparece en el periodo agudo, en el 100% de los casos, y suele ser elevada, a veces de 40 °C. No responde a los antibióticos y sí de forma parcial a los antitérmicos. El periodo febril puede durar de 5 a 25 días con un tiempo medio de 10 días.
• Conjuntivitis: ocurre en el 85% de los casos, y se trata de una inyección conjuntival no supurativa bilateral, frecuentemente intensa, principalmente en la conjuntiva bulbar. En algunos casos puede asociarse a uveítis anterior transitoria y benigna.
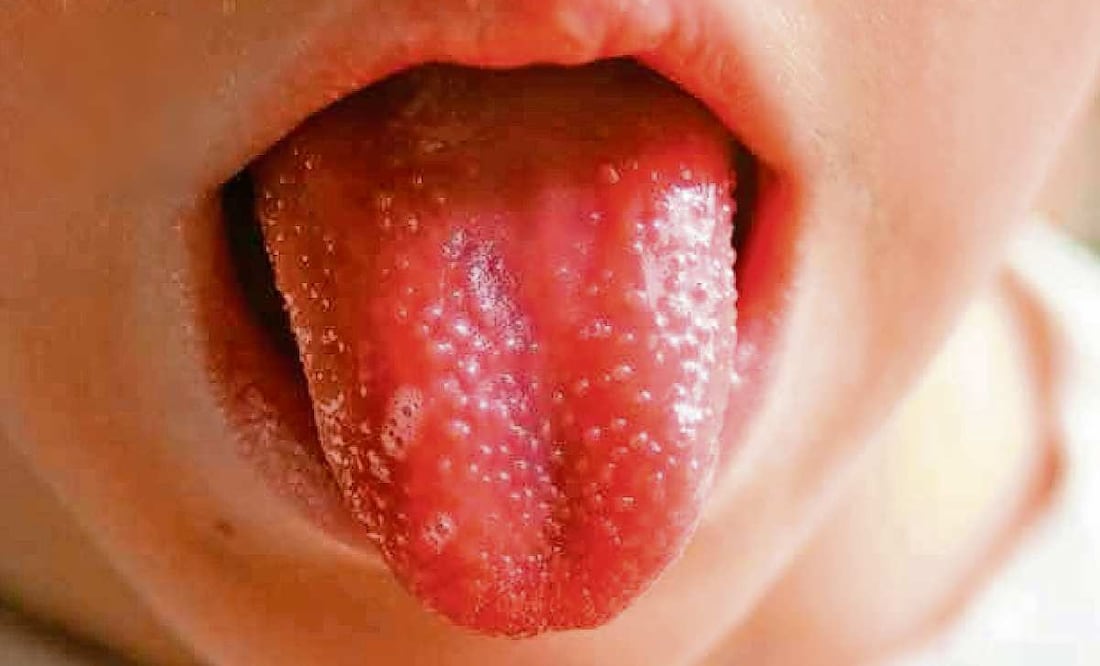
• Alteraciones bucales: aparecen en el 90% de los casos, se inician en la fase aguda, y duran aproximadamente lo mismo que las lesiones oculares. Los labios pueden estar secos, agrietados, así como engrosados, con una lengua de aspecto aframbuesado. Puede haber enantema.
• Exantema: es polimorfo, y puede ser maculo-papuloso, escarlatiniforme o multiforme. Comienza frecuentemente en superficies extensoras de los miembros, para después extenderse por el tronco. Es característica la afectación perineal. No se aprecian vesículas, costras ni bullas, como las observadas en el síndrome de Stevens-Johnson. La erupción suele desaparecer en 1 o 2 semanas, al igual que la fiebre.
• Alteraciones en las extremidades:
– Inicial: se trata de un eritema florido de las palmas de las manos y las plantas de los pies asociado, o no, a un edema indurado, sin impresión digital
.– Fase de convalecencia: al final de la segunda semana se produce una descamación de comienzo a nivel periungueal, característica de la enfermedad, aunque puede no presentarse. En la fase de convalecencia pueden presentarse también las líneas de Beau (líneas profundas transversas que atraviesan las uñas).
• Linfadenopatías: es el criterio mayor que aparece en menos ocasiones, aproximadamente en un 70%. La linfadenopatía cervical suele ser unilateral, con afectación de un único ganglio linfático, doloroso, duro y de más de 1,5 cm de diámetro, que remite a medida que cede la fiebre. Ocasionalmente puede haber adenopatías más generalizadas.
• Afectación cardiaca: esta característica debe considerarse como sinónimo de la enfermedad y siempre se debe pensar que puede estar presente, se debe buscar, y vigilar, porque es la afectación que puede otorgarle gravedad al proceso. Las manifestaciones cardiacas son muy variables: desde alteraciones inespecíficas del electrocardiograma (ECG) sin repercusión clínica, hasta soplos cardiacos, ritmo de galope, pericarditis, endocarditis, miocarditis y aneurismas coronarios. En los primeros días del proceso, puede detectarse arteritis de la coronaria. Pasado el primer mes es cuando se pueden detectar los aneurismas y las trombosis, por ecocardiografía o por técnicas angiográficas. La mayoría de los aneurismas regresan espontáneamente en un año. La tasa de mortalidad está entre un 1 y un 2‰.
• pueden aparecer otros síntomas y signos clínicos en el curso de la enfermedad, sobre todo en la fase febril aguda. Los pacientes pueden presentar irritabilidad importante que puede ser secundaria a una meningitis aséptica o resultado de una lesión de tipo vasculítica focal neurológica. También pueden presentar tos, afonía, mucosidad u otros síntomas sugestivos de un cuadro de infección vírica del tracto respiratorio. En ocasiones van acompañados de infiltrados pulmonares u otitis media. Puede aparecer dolor abdominal (similar al observado en los niños con púrpura de SchönleinHenoch), acompañado o no de diarrea. Otros síntomas menos frecuentes son: elevación de las transaminasas, ictericia, hidrops de vesícula biliar, artralgias o artritis (un 55% de ellas oligoarticular, 45% poliarticular principalmente en rodillas, codos y grandes articulaciones). En algunos casos puede producirse un cuadro de hipertensión intracraneal con papiledema bilateral secundario. Se han descrito también casos de hipoacusia, generalmente reversible.
El diagnóstico de la enfermedad de Kawasaki es clínico debido a la ausencia de pruebas específicas, y se basa en la identificación de los principales hallazgos clínicos como ya se ha mencionado y la exclusión de otras entidades de causas conocidas. En casos de retraso en el diagnóstico y tratamiento, o en casos refractarios, se pueden formar aneurismas que conducen a secuelas severas como infartos cardíacos. Globalmente es considerada la vasculitis primaria más común durante la niñez, y en Europa central y Norteamérica es la segunda más frecuente después de la púrpura de HenochSchönlein; además, se considera la condición cardiaca adquirida más común en países desarrollados.
Esta patología fue identificada por primera vez en una publicación por el Dr. Tomisaku Kawasaki de 1967, en la que se documentaron 50 pacientes pediátricos con vasculitis registrados desde 1961. Se describe como un síndrome mucocutáneo febril con compromiso linfoide y descamación específica de dedos de la mano y el pie, lo cual se mantiene prácticamente sin cambios hasta la actualidad. después de esta primera publicación, existió controversia significativa en cuanto a la relación del complejo de signos y síntomas descritos por Kawasaki y las complicaciones cardiacas presentes en estos pacientes. Esta controversia fue resuelta en 1970 cuando se documentaron, por hallazgos histopatológicos, diez casos de muerte cardiaca súbita en pacientes con enfermedad de Kawasaki.
El Dr. Kawasaki realizó su primera publicación en inglés en 1974 y, para entonces, la relación entre la patología y la vasculitis de arterias coronarias estaba bien establecida. La consecuencia más temida de la enfermedad de Kawasaki es el aneurisma de arterias coronarias, y su incidencia se puede reducir drásticamente con un diagnóstico certero y un tratamiento oportuno con inmunoglobulina intravenosa y aspirina.
Para el diagnóstico de la enfermedad es necesario considerar que puede haber cuadros clínicos de presentación parcial y de presentación completa con todos los datos que hemos mencionado como propios de la enfermedad, se dice que se encuentra la enfermedad completa cuando se identifican:
Son necesarios la presencia de fiebre durante más de 5 días junto con 4 criterios clínicos o bien la fiebre junto con 3 criterios adicionales que se identifican si el paciente presenta afectación cardiaca compatible.
En presencia de ≥4 criterios principales, se puede diagnosticar de EK al cuarto día de en Fiebre >5 días y al menos 4 de los siguientes criterios: - Inyección conjuntival bilateral
- Alteraciones de las mucosas labiales o faríngeas. Enantema, lengua aframbuesada y/o labios fisurados
• Cambios periféricos de las extremidades, que incluyen, edema, eritema y/o descamación (puede ocurrir más tarde)
• Rash o exantema polimorfo
• Linfadenopatía cervical de más de 1,5 cm
hallazgos clínicos que ayudan al diagnóstico - Leucocitosis >15 000 con desviación a la izquierda
• Anemia normocítica normocrómica
• Trombocitosis >450 000
• PCR >40 mg/dl
• VSG >40 mm/h
• Elevación ALT, AST, GGT y ferritina
• Albúmina <3 g/dl Sedimento urinario con piuria estéril (>10 leucocitos/campo con urocultivo negativo)
Faringotest negativo
Exploración oftalmológica con signos de uveítis anterior
Ecografía abdominal con hidrops de vesícula biliar
Ecocardiografía: - Miocarditis
• Insuficiencia mitral
• Alteraciones del ritmo cardíaco
• Aneurismas
• Vasculitis coronaria
Clínica acompañante
• Artralgias/artritis reactiva
• Meningitis aséptica
• Irritabilidad
• Exantema de inicio a nivel perineal
• Uveítis anterior
• Meatitis y disuria
• Hidrops de vesícula biliar/colestasis
• Ictericia/hepatitis









