
Dr. Guillermo Flores Flores/Diario de Chiapas
Enfermedad Glomerular de Cambios Mínimos
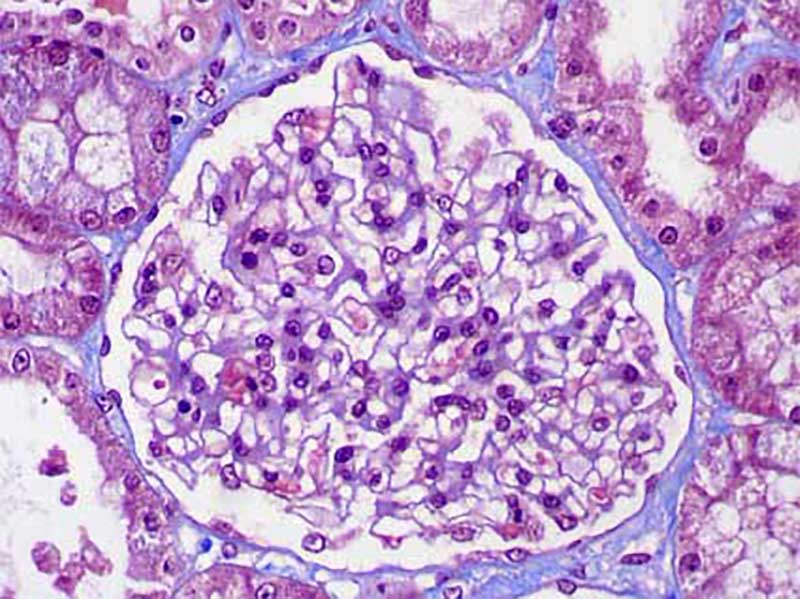
El síndrome nefrótico asociado a cambios glomerulares mínimos (Glomeruloesclerosis de cambios mínimos) es una enfermedad de causa desconocida, que afecta predominantemente a niños, y en la que no hay alteraciones histológicas glomerulares evidenciables con la microscopía de luz convencional, o éstas son sutiles. Su diagnóstico se confirma por la demostración de pérdida (o borramiento o simplificación) de los procesos podocitarios (también se le conoce como fusión de procesos podocitarios). La inmunofluorescencia es típicamente negativa.
La enfermedad ha recibido varios nombres: nefropatía de cambios mínimos, nefrosis lipoidea, enfermedad de los procesos podocitarios, “enfermedad con hallazgos histopatológicos nulos” (una traducción aproximada de “nil disease”), enfermedad epitelial glomerular, etcétera. El término nefrosis lipoidea fue utilizado antes del uso generalizado de la biopsia renal y sugería una alteración en el metabolismo de los lípidos; la razón para usarlo fue que en estos pacientes se ven cuerpos grasos en la orina y cambio graso en algunas células tubulares en la autopsia.
Hay formas primaria (idiopática) y secundaria de la enfermedad. Estas formas secundarias pueden ser el resultado de tratamiento con antiinflamatorios no esteroideos, enfermedades linfoproliferativas (más frecuentemente enfermedad de Hodgkin) y reacciones alérgicas (como a picaduras de abeja).
Es difícil determinar la incidencia exacta de la enfermedad debido a que en niños con síndrome nefrótico no se suele hacer biopsia, a menos que no respondan a esteroides o que sea corticorresistente. Se calcula que 60% a 70% de los niños con síndrome nefrótico tendrán CGM. Es, por lo tanto, la causa más frecuente de síndrome nefrótico en este grupo etario. Es más frecuente en niños que en niñas: 2:1, y la edad promedio de presentación está entre los 3 y 4 años de edad; el 80% de los niños está por debajo de los 6 años. En adultos corresponde al 10-20% de pacientes con síndrome nefrótico y en esta edad no hay la clara predominancia masculina.
Contrario a la glomeruloesclerosis focal y segmentaria (Glomeruloesclerosis Esclerosante Focal y Segmentaria), la enfermedad de CGM ocurre más frecuentemente en caucásicos que en afroamericanos.
El pronóstico es relativamente bueno, usualmente responden al tratamiento con corticosteroides con desaparición de las lesiones ultraestructurales. En adultos con síndrome nefrótico se debe hacer biopsia; en niños, sólo si son corticorresistentes, corticodependientes o hay hallazgos que hagan sospechar una enfermedad diferente.
La causa de la glomeruloesclerosis de cambios mínimos es desconocida. Aunque no hay depósitos inmunes en los glomérulos de estos pacientes, se ha sospechado de un mecanismo inmune como causa de la enfermedad. En muchos niños la alteración está asociada a una historia de atopia, alergias alimentarias y reacciones de hipersensibilidad. Algunos autores en 1958, se refirieron a la Glomeruloesclerosis der cambios Mínimos como “asma de la nefrona”. Se han descrito numerosas alteraciones de la inmunidad humoral y celular en los afectados por esta alteración. Hay muy poca evidencia de una respuesta humoral como causa, en cambio, recientemente ha recibido más atención la respuesta celular. La respuesta a esteroides y ciclofosfamida en la mayoría de estos pacientes es un fuerte indicio de que la inmunidad celular tiene un importante papel en la patogénesis. Probablemente linfoquinas secretadas por algún tipo de células T tengan capacidad para producir, directa o indirectamente, los cambios estructurales y funcionales característicos.
Tampoco está completamente claro cómo la alteración estructural en los podocitos lleva a la proteinuria. En algunos trabajos se ha encontrado reducción en la carga iónica de la pared capilar, lo que facilitaría el transporte de proteínas a través de la barrera glomerular.
Un gran avance en el entendimiento del síndrome nefrótico idiopático, y por ende de CGM y de Glomerulonefritis Esclerosante Focal y Segmentaria, se produjo con el descubrimiento, en 1998, de la nefrina (NPHS1), el gen mutado en el síndrome nefrótico congénito de tipo finlandés; fue crucial para establecer que el podocito es el componente central de la barrera de filtración. Poco después se descubrió el gene mutado en el síndrome nefrótico corticorresistente de inicio temprano: NPHS2 que codifica otra proteína exclusiva del podocito: la podocina. In vivo sabemos que la nefrina está ubicada en el diafragma de hendidura (“slit diafragma”) e interactúa con podocina con una unión a la proteína asociada a CD2 (CD2AP). Esta última se conocía como una molécula ligada al citoesqueleto de células T. La podocina y nefrina se unen al citoesqueleto del podocito (proteínas transmembrana) y su disrupción no sólo afecta el diafragma de hendidura sino la integridad de todo el proceso podocitario. Aunque muy poco claro aún, la fisiopatogenia del síndrome nefrótico en CGM y Glomerulonefritis Esclerosante Focal y Segmentaria afecta este complejo, ya sea, como en algunos casos, por defectos genéticos en estas proteínas (hay ya muchos descritos) o por lesión secundaria a un(os) mediador(es) inmune(s) o no inmune(s). En el capítulo de Glomerulonefritis Esclerosante Focal y Segmentaria profundizaremos un poco más acerca de este complejo que forman el diafragma y los procesos podocitarios, claves para entender la enfermedad y centro actual de la investigación de primera línea en estas enfermedades
Clínica: La principal manifestación es proteinuria, usualmente en rango nefrótico, con edema periorbitario. En muchos casos hay las otras manifestaciones del síndrome nefrótico, pero, pueden estar ausentes. Es inusual la hematuria y cuando se encuentra es microscópica. La proteinuria es selectiva, a diferencia de la proteinuria en Glomerulonefritis Esclerosante Focal y Segmentaria y en Glomérulo Nefritis membranosa (esto se determina por electroforesis de proteínas en orina, aunque pocas veces se utiliza esta prueba). Hay informes de casos de CGM con proteinuria muy leve y/o hematuria como únicos hallazgos, sin embargo no está claro si se trata de una “forma frustra” de la enfermedad o, como lo proponen algunos autores, “cambios glomerulares inespecíficos”. No suele encontrarse alteración en la función renal.
Las manifestaciones clínicas en adultos son similares a las de los niños, sin embargo, hay una incidencia más alta de hipertensión, falla renal y proteinuria no selectiva.
La respuesta a esteroides es alta: más del 90% en niños. Muchos de estos pacientes presentan recaídas que, en general, responden también al tratamiento. Las recaídas tienden a desaparecer antes de llegar a la edad adulta. Los porcentajes de pacientes con remisión completa, recaídas, dependencia de esteroides y no respuesta al tratamiento son muy variables en la literatura. Alrededor del 5% continúa presentando recaídas en la edad adulta y menos del 3% llegan a falla renal a los 10 años del diagnóstico. En los pacientes cortico-resistentes, a menudo, se utilizan medicamentos citotóxicos como ciclofosfamida.
En adultos la respuesta a esteroides es menor: 60-90% de casos. Además, suele haber un mayor intervalo de tiempo entre el inicio del tratamiento y la respuesta clínica.
En las formas secundarias el pronóstico depende de la condición subyacente. La enfermedad tiende a desaparecer al resolverse la causa asociada.
Entre los factores pronósticos adversos se han sugerido: frecuencia de recaídas, proteinuria no selectiva, alteración de la función renal e hipercelularidad mesangial.
Datos de laboratorio: Proteinuria selectiva: albumina y otras proteínas de bajo peso molecular. La presencia de proteínas de alto peso sugiere otro diagnóstico. Hematuria microscópica en 10-30%. BUN y creatinina usualmente normales. C3, C4 y otras pruebas que indiquen formación de complejos inmunes son normales.
Histopatología
Por definición no hay, o son mínimos, cambios histológicos glomerulares. Como en cualquier síndrome nefrótico pueden evidenciarse gotas de reabsorción proteica en el citoplasma de podocitos y estas células pueden aparecer prominentes. La celularidad es normal. En algunos casos hay hipercelularidad mesangial leve; es muy discutido si este hallazgo se correlaciona con una menor respuesta a esteroides, algunos trabajos no han encontrado tal relación. Cuando hay hipercelularidad es más probable encontrar depósitos de IgM (nefropatía IgM, ver más adelante). No hay lesiones glomerulares segmentarias; si las hubiere no debe diagnosticarse Glomeruloesclerosis de Cambios Mínimos.
Pueden encontrarse algunos glomérulos globalmente esclerosados, como en cualquier individuo normal; la fórmula tradicionalmente usada para determinar el máximo permitido de glomeruloesclerosis global es: edad, dividido por 2, menos 10; esta fórmula se aplica a adultos; en niños no debemos esperar más que un ocasional glomérulo esclerosado. Cualquier lesión segmentaria, por sutil que sea, debe obligarnos a plantear otro diagnóstico, como Glomerulonefritis Esclerosante Focal y Segmentaria.
Inmunofluorescencia
Lo habitual es negatividad para inmunoglobulinas y fracciones del complemento. En algunos casos hay depósitos débiles de C3 en el mesangio, sin que esto se correlacione con la presentación clínica o la evolución. Cuando hay depósitos mesangiales intensos de IgM o de C1q probablemente hay implicacione importantes en la patogénesis y evolución de la enfermedad.
Microscopía
electrónica
Los cambios ultraestructurales diagnósticos están entre el podocito y la MBG. La arquitectura de los procesos podocitarios está perdida, no hay hendiduras o diafragmas de filtración. A este hallazgo se le llama simplificación, fusión o borramiento de los procesos podocitarios. El citoplasma de podocitos presenta prolongaciones largas y estrechas desde la superficie: “transformación microvellosa”. Es frecuente que el citoplasma se vea más electro-denso en la parte adyacente a la membrana basal glomerular: condensación del citoesqueleto. La alteración de podocitos suele ser difusa, sin embargo, hay casos en los que el hallazgo es focal. No hay una correlación constante entre el grado de lesión ultraestructural y la severidad de la proteinuria. Estos cambios revierten cuando hay respuesta al tratamiento.
El tratamiento farmacológico de la Nefropatia de Cambios Minimos se centra en provocar la remisión para restaurar la función glomerular e invertir los síntomas y signos del síndrome nefrótico. Este tratamiento consiste principalmente en corticoesteroides, como la prednisona o la prednisolona, para lograr la inmunodepresión y reducir la actividad de las citosinas de los linfocitos T9. Aunque los niños generalmente responden a los corticoesteroides en un período de 2 semanas y continúan tomándolos durante otras 6 semanas, los adultos responden mucho más lentamente y pueden llegar a tomarlos durante 16 semanas. Después de iniciar el tratamiento, remiten los síntomas del 80-95% de los pacientes. Además, hasta el 20% de los pacientes pueden desarrollar resistencia a los corticoesteroides. Los medicamentos inmunodepresores, como la ciclosporina, se pueden utilizar para tratar la resistencia a los corticoesteroides.









